
Resources: Blog
I used to think RNA was the phenotype
Gary Schroth, CSO
May 6, 2026
I used to think that if we could sequence everything, genomic DNA coupled with RNA from every cell, we could figure out everything. That assumption shaped most of my career.
It was not an unreasonable place to start. DNA was the blueprint. But every cell in your body carries essentially the same genome. A neuron and a macrophage have the same DNA. What makes them different is not their genome but what they do with it. The genome alone was never going to explain everything. We needed to read the transcriptome.
The transcriptome turned out to be far richer than anyone expected. One of my first RNA-Seq studies, published in Nature in 2008, applied the technology across 15 diverse human tissues and cell lines. We found thousands of previously unknown splice sites and showed that approximately 94% of human genes undergo alternative splicing. The diversity of what the transcriptome was doing, tissue by tissue, was astonishing. It went on to become the most-cited paper of my career. It felt like the field had found the right readout at last.
From bulk tissue body maps, the field moved to single cells. RNA-Seq revealed that populations we thought were homogenous were actually dozens of distinct states. Cell atlases of the brain, the gut, the immune system mapped a level of cellular diversity nobody had fully appreciated before. The Human Cell Atlas. Whole-brain maps. Immune system atlases. Remarkable catalogs of what cells are.
And yet atlases are portraits, not films. They tell you what a cell is. They do not tell you what it is going to do.
I remember reading Lewis Thomas's "The Lives of a Cell" in high school biology, and it had a huge impact on my interest in science generally, but specifically in biology. Thomas wrote about cells with genuine astonishment at what they actually do. He was not interested in the parts list. He was interested in behavior: how cells move, signal, respond, fail, recover, and coordinate with everything around them. He was also quietly suspicious of any claim that the hard problem of living systems had been solved.
Then I spent the next thirty years learning to read the molecular programs of cells. And at some point, the program became the thing. The transcriptome became the cell.
Two preprints we posted to bioRxiv this week showed me precisely where that assumption breaks.
In work on preadipocytes during adipogenesis, we measured lipid accumulation directly by imaging and read the transcriptome from the same cells. When we clustered the transcriptomic data the standard way, cells with high and low lipid accumulation did not separate cleanly. The dominant axes of transcriptional variation had little to do with the function we were measuring.
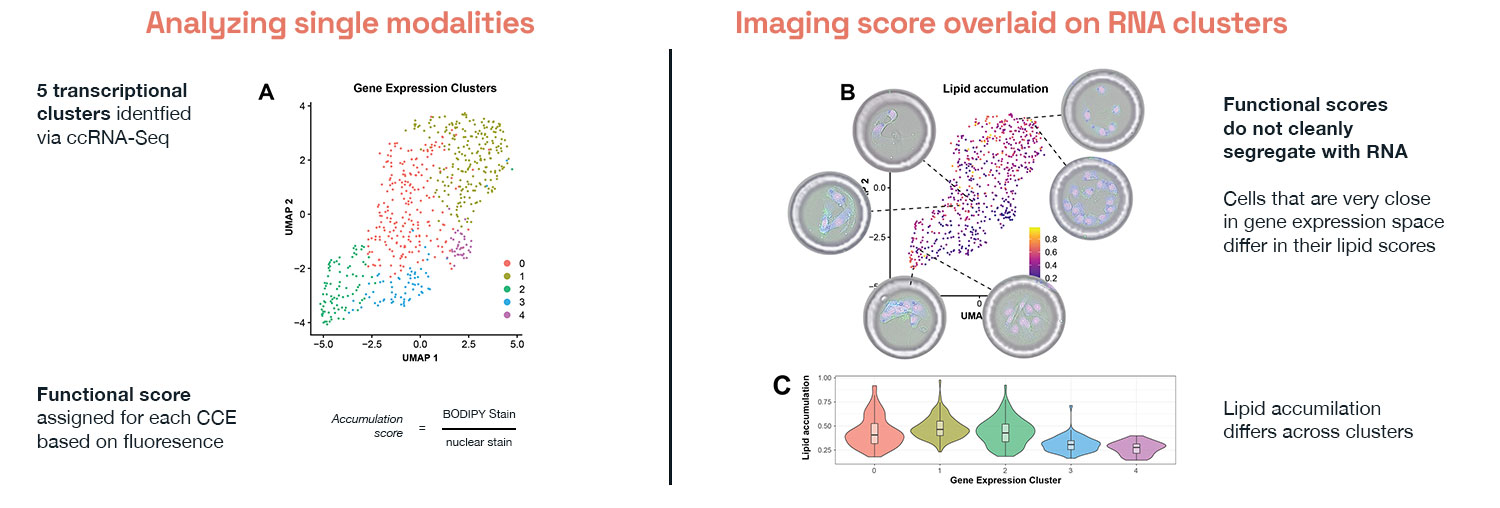
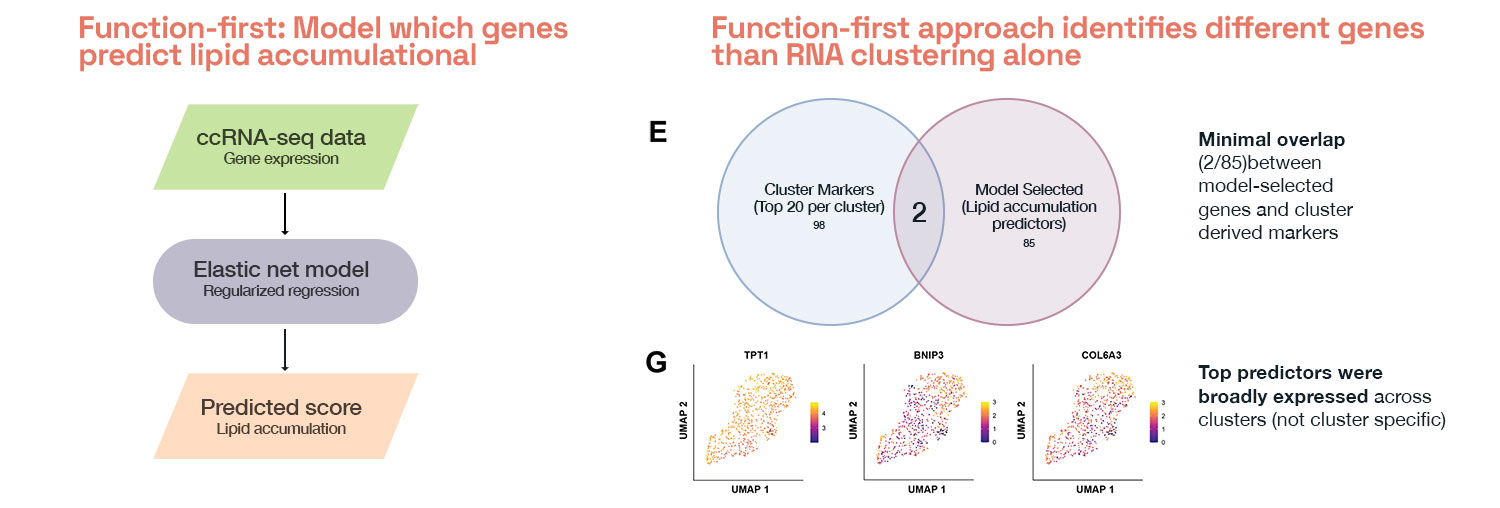
So we flipped the question. Because measurements were linked in the same cells, we could work backwards from phenotype to find the genes that drive it. We could ask ‘Which genes predict the outcome we care about?’ and ‘How does that compare to the genes nominated by cluster analysis?’
When we trained a model to predict lipid accumulation from gene expression directly, it selected 85 predictor genes. Fewer than 10% overlapped with the top cluster-defining genes. Same cells, same RNA data, different question, completely different gene list.
The same pattern appeared in microglial phagocytosis. Morphology alone outperformed gene expression alone in predicting phagocytic activity. The top functional predictors, Gpnmb and Clec4e, have known mechanistic roles in phagocytosis, and yet they were not among the top cluster markers. The transcriptomic clusters were real. They were just answering a different question than the one we were asking.
The lung cancer result changed how I think about resistance. We treated A549 cells with Olmutinib, an EGFR inhibitor, and tracked individual cells over time by imaging. One class we identified were daughter-cell resistant: a cell divided, one sibling died while the other survived. These were not a pre-existing resistant clone. They were siblings, and one of them landed in a gene expression state that happened to be protective, characterized by potassium channel upregulation and p53-dependent quiescence. Longitudinal imaging told that story. A transcriptomic snapshot of the population would have buried it in noise.
What these results point to is a definitional question I had stopped asking.
Classical genetics used phenotype to mean what an organism or cell does, its observable behavior. The single-cell sequencing era compressed that definition. Transcriptional state became a shorthand for phenotype, and for questions about cellular identity, it worked well enough that the compression felt harmless. What is this cell? RNA answers that question well.
But the questions the field is now asking are often different. What will this cell do when you add a drug? Which cell will resist, differentiate, kill, phagocytose? Which clone survives? Which cell in this interacting pair is driving the outcome? For those questions, phenotype has to mean behavior again, with molecular state attached.
This is not an argument against RNA. RNA paired with observed behavior in the same cells is a more informative readout than RNA alone. The platform we built at Cellanome links live-cell imaging with whole-transcriptome capture from the same individual cells, and that pairing is what made these analyses possible. But the design principle applies more broadly. If the biological question you are asking is a function question, ask yourself whether a transcriptome-only readout can answer it. If the answer is no, or not cleanly, the experiment needs a functional observation alongside the RNA.
Thomas wrote those essays when molecular biology was just beginning to reveal what was inside the cell. He already knew that what was inside was not the point. His argument, at its core, was for seeing life whole: as a web of relationships and behaviors rather than a collection of isolated parts. What the cell did, how it communicated and responded in relation to other cells over time, that was the biology worth watching. He was also comfortable not knowing. Distrustful, even, of fields that had grown too confident about what they had explained.
He was right. It took me a while to fully come around to it.
— GS
Next up, I will apply this argument to a place where it has real practical weight: pooled CRISPR screens, where the readout determines the hit list.
